Variant calling using Sniffles 
Despite its error rate recently released tools such as Sniffles have shown the ability to call structural variants using noisy long-read data.
Here, we will use minimap2 and Sniffles to call variants on the provided data and visualise it using the Integrative Genome Browser (IGV).
First, change into the variant_calling_practical directory. Concatenate all fastq files from the ~/course_data/precompiled/guppy_output/ into one fastq file and use minimap2 to map them to the reference.
cd ~/course_data/practicals/variant_calling_practical
cat ~/course_data/precompiled/guppy_out/*.fastq > ./all_guppy.fastq
minimap2 --MD -a ~/course_data/prcompiled/chr17 \
~/course_data/practicals/basecalling_practical/all_guppy.fastq > mapped.sam
The additional minimap2 options in the above command will write the output in SAM in stead of PAF format (option –a) and also include the MD-tag in the annotation needed by sniffles (–MD).
Before we can use the mapped file we have to do real bioinformatics: file conversion!
 Use the tool samtools to
Use the tool samtools to
- Convert the sam file to bam (a binary sam format) using samtools’ view command
- Sort the bam file (needed for fast access) using samtools sort command
- Create an index of the bam file (needed by IGV) using samtools index command
# Convert to bam file
samtools view –bS mapped.sam > mapped.bam
# Sort the bam file
samtools sort –o mapped.sorted.bam mapped.bam
# create an index file
samtools index mapped.sorted.bam
Run sniffles on the sorted bam file to write a out a Variant Call Format (vcf) file.
sniffles -m mapped.sorted.bam -v variants.vcf
To visualise the sniffles results we will use IGV:
- Type igv on the command line to open IGV
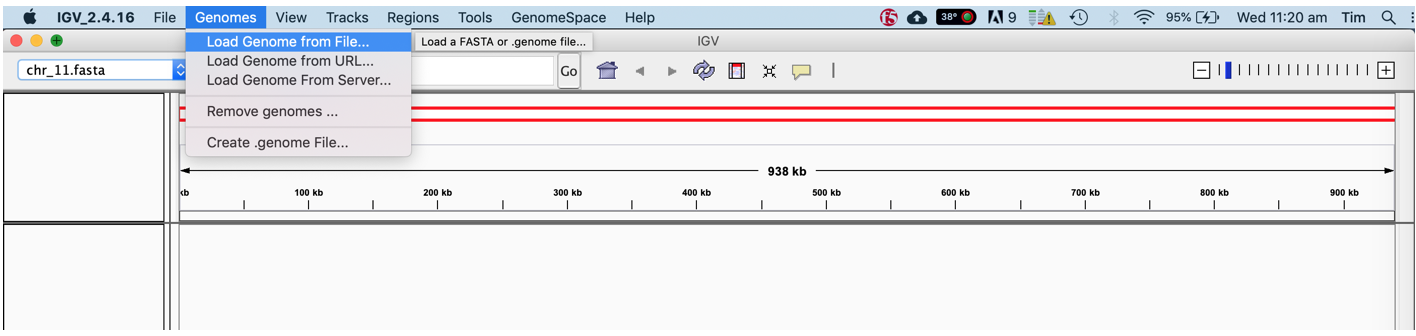
- Click the Genomes -> Load Genome menu and open the chr17.fasta in the precompiled folder
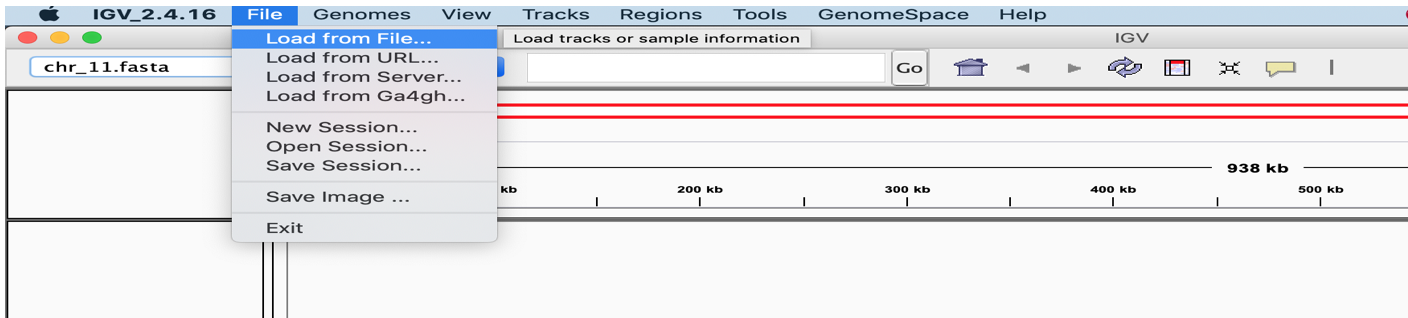
- Use the File -> Load from File menu to load variants.vcf and the mapped.sorted.bam files you just created
Your screen should now look something like this
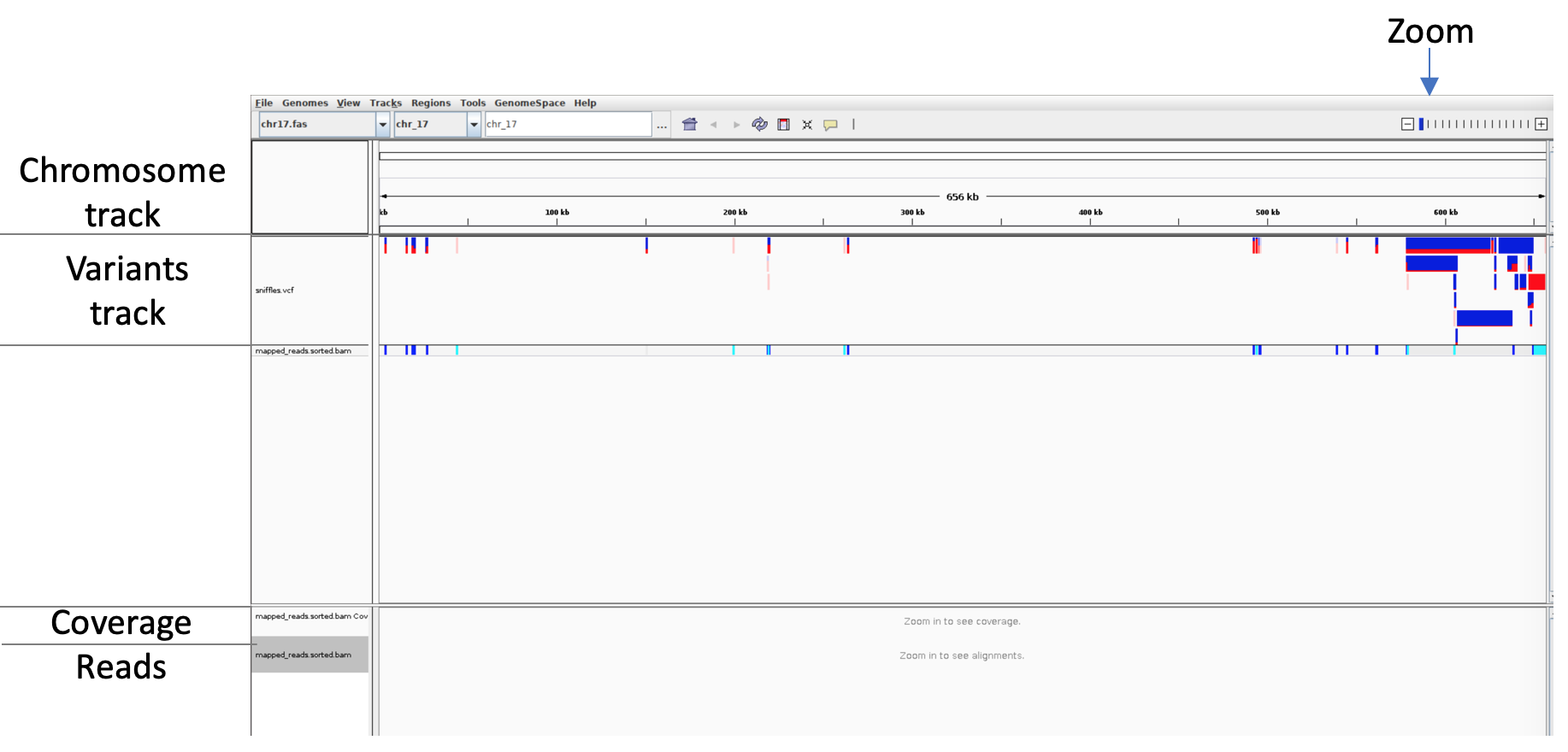
Get familiar with IGV. Things to try are
- Zoom in until you see the reads mapped to the reference, Right-click on the bam-track and select “Collapse” and “link supplementary alignments”
- Hover over a variant to see a description of the variant
- What type are the majority of the variants?
- Is there anything weird going on with the coverage at variant positions? What could be the explanation?